Erfahrungen und Fallbeispiele
Erfahrungen und Fallbeispiele
Die Pharmaindustrie zieht seit vielen Jahren das Interesse von Kapitalinvestoren auf sich. In letzter Zeit hat das chinesische Pharma-F&E-Segment sowohl bei inländischen als auch globalen Investoren zunehmend Aufmerksamkeit erregt. Im Jahr 2020 hat Herr Xiang Tang (Rechtsanwalt) erfolgreich einen Fall betreut, bei dem sein (internationaler) Kunde beabsichtigte, Anteile an einem innovativen chinesischen Pharma-F&E-Unternehmen (dem "Zielunternehmen") zu erwerben. Herr Tang führte die Verhandlungen in diesem Deal und brachte dabei seine Fachexpertise ein, einschließlich der Ausarbeitung der Absichtserklärung, Durchführung der rechtlichen Due Diligence, Unterstützung bei den Verhandlungen des Mandanten und Koordination mit anderen externen Beratern. Derzeit werden Preisverhandlungen zwischen dem Kunden und dem Zielunternehmen geführt.
Das Hauptgeschäft des Zielunternehmens besteht in der unabhängigen F&E innovativer Medikamente zur Behandlung von Tumoren, darunter Lungenkrebs, Gliom und Brustkrebs, mit einem besonderen Schwerpunkt auf der Entwicklung von zielgerichteten kleinen Molekülen für Hirntumoren, die klinisch selten sind. Aufgrund der einzigartigen Merkmale der Pharmaindustrie liegt der Fokus des gesamten rechtlichen Due-Diligence-Prozesses auf der Untersuchung und Analyse der Qualifikationen und des Geschäftsmodells des Zielunternehmens.
Eine Übersicht über das Geschäftsmodell des Zielunternehmens finden Sie unten:
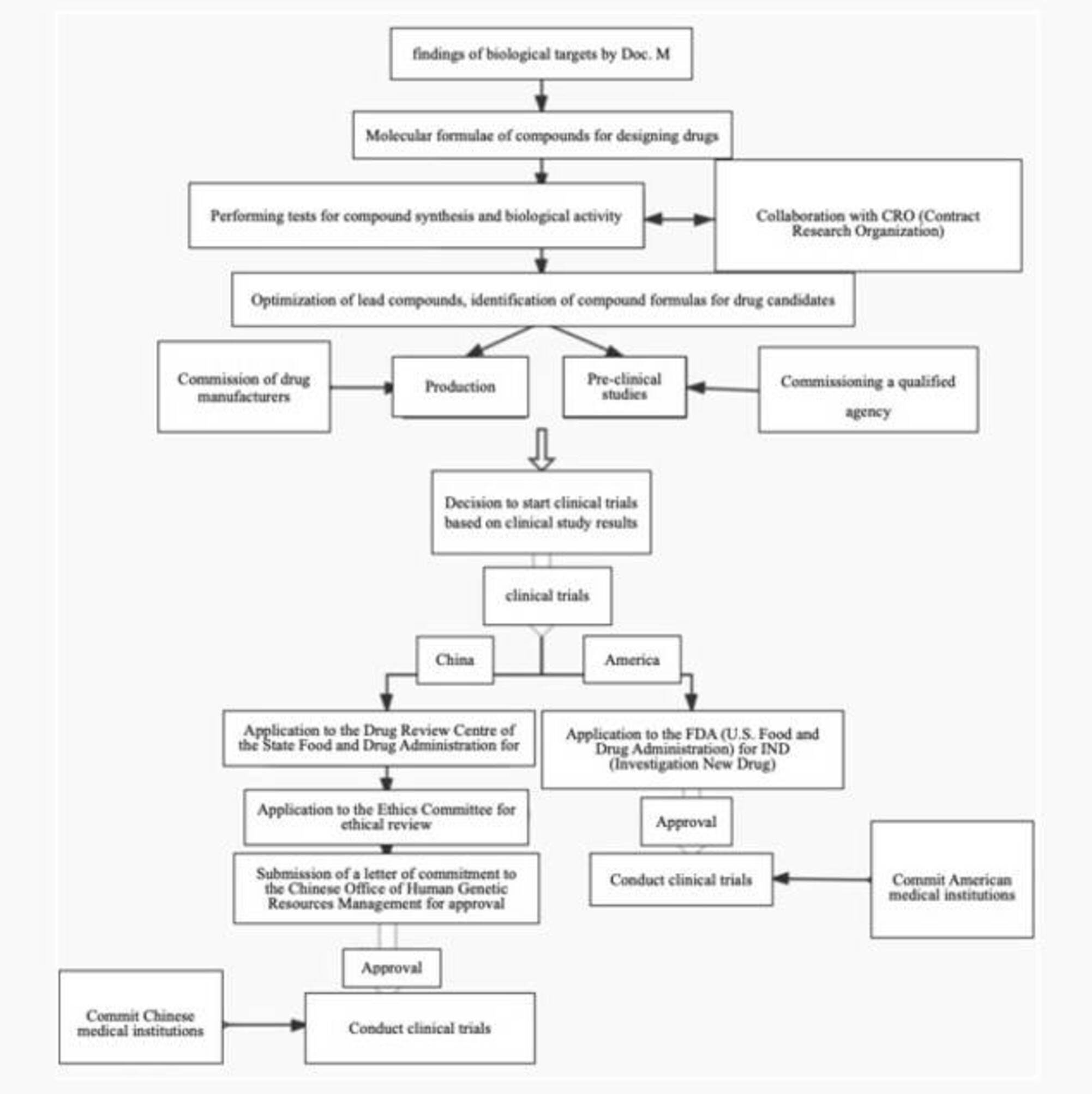
Nach Klärung des Geschäftsmodells des Zielunternehmens ist es notwendig, weiter zu überprüfen, ob es über die relevanten Qualifikationen verfügt und die entsprechenden Verpflichtungen erfüllt hat. Im aktuellen Arzneimittelzulassungsverfahren muss ein Medikament den Weg von präklinischen Studien über klinische Versuche bis zur Vermarktung gehen. Hierbei durchläuft es die Phasen der präklinischen Studien, einschließlich der Entdeckung und Bestätigung von Arzneimittelzielen, der Synthese und Screening von Verbindungen, pharmakologischer Forschung, pharmakologischer und toxikologischer Forschung sowie der Entwicklung von Formulierungen, etc. Die klinischen Versuche umfassen Phase I, II und III. Die aktuellen Gesetze und Vorschriften in China enthalten spezifische Bestimmungen für die oben genannten verschiedenen Stadien, beispielsweise:
Präklinische Forschung
- Die Auftraggeberpartei ist für die Studienmaterialien verantwortlich, die für die Anmeldung [1] eingereicht werden, und der Inhaber der Arzneimittelzulassung ist verantwortlich für die nicht-klinische Studie des Arzneimittels[2]. Die Forschungseinrichtung muss sich an die einschlägigen Bestimmungen der Guten Laborpraxis für nicht-klinische Labortests halten.
- Für Einheiten, die tatsächlich an Studien zur nicht-klinischen Sicherheitsbewertung von Arzneimitteln beteiligt sind, sollten diese Studien an Einrichtungen durchgeführt werden, die nach den Grundsätzen der Guten Laborpraxis für nicht-klinische Labortests zertifiziert sind [3].
Klinische Studien
- Die klinischen Prüfeinrichtungen für Arzneimittel werden durch Einreichung verwaltet, und Antragsteller sollten Arzneimittelprüfeinrichtungen auswählen, die in der "Plattform für das Management der Einreichung von Arzneimittelprüfeinrichtungen" eingereicht wurden, um Arzneimittelklinische Studien durchzuführen. [4]
- Der Inhaber einer Arzneimittelzulassung ist für die klinischen Studien des Arzneimittels verantwortlich. [5]
Neuzulassung für die Vermarktung eines neuen Arzneimittels
- Um in China vermarktet zu werden, müssen Arzneimittel von der zuständigen Behörde des Staatsrats für Arzneimittelüberwachung und -verwaltung genehmigt und eine Arzneimittelzulassung erhalten; mit Ausnahme von chinesischen Kräutermedizin und chinesischen Arzneimitteltabletten, die keiner Genehmigung und Verwaltung unterliegen. [6]
- Der Inhaber einer Arzneimittelzulassung ist gemäß dem Gesetz für die Sicherheit, Wirksamkeit und Qualitätskontrolle des Arzneimittels während des gesamten Prozesses der Arzneimittelentwicklung, -produktion, -betrieb und -nutzung verantwortlich. Der gesetzliche Vertreter und die verantwortliche Person des Inhabers einer Arzneimittelzulassung sind vollumfänglich für die Qualität des Arzneimittelprodukts verantwortlich. [7]
Die Erforschung und Entwicklung neuer Medikamente ist eine Industrie mit hohem Risiko, hohen Investitionen und gleichzeitig hohen Ertragsaussichten. Sie ist durch eine längere Startphase, beträchtliche Investitionen und eine geringe Erfolgsquote gekennzeichnet, erfordert jedoch gleichzeitig strenge Überwachung und Management. Daher erfordert die Bewertung von Unternehmen im Bereich der Forschung und Entwicklung neuer Medikamente eine umfassende, vielschichtige Analyse auf verschiedenen Ebenen, was diese Angelegenheit zu einer komplexen Herausforderung macht.
- Artikel 49 der Grundsätze der Guten Laborpraxis für nicht-klinische Labortests.
- Artikel 30 des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China.
- Artikel 10 der Maßnahmen zur Verwaltung der Arzneimittelzulassung, herausgegeben von der Staatlichen Lebensmittel- und Arzneimittelverwaltung.
- Artikel 19 des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China und Artikel 30 der Verordnung zur Durchführung des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China.
- Artikel 30 des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China.
- Artikel 24 des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China.
- Artikel 6 und Artikel 30 des Gesetzes über die Verwaltung von Arzneimitteln der Volksrepublik China.